圖1.四種可能吸附構型的分布及結構圖示
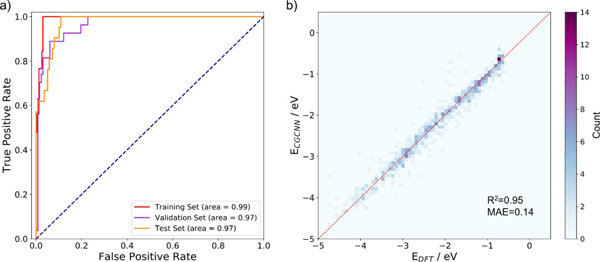
圖2.機器學習模型的性能。a)分類性能;b)回歸性能

圖3.多硫化鋰吸附能及過電勢的預測。a)LiS*的吸附能;b)不同催化劑上最弱的可溶性多硫化鋰吸附能;c)所預測的全部催化劑的“火山圖”;d)過電勢低于0.1V的催化劑;e)預測的過電勢。
近日,中國科學院金屬研究所沈陽材料科學國家研究中心聯合研究部副研究員李波研究小組,采用基于高通量密度泛函理論計算的機器學習方法,系統研究了多硫化物的吸附模式,并對上千種氮摻雜碳材料負載的過渡金屬單原子催化劑進行了篩選,為鋰硫電池正極材料中單原子催化劑的設計提供了指導。
研究人員首先采用密度泛函理論對800余個吸附結構進行計算,結果顯示多硫化鋰在催化劑上有四種可能的吸附構型,并可分為兩大類,即解離吸附和非解離吸附。基于晶體圖卷積神經網絡訓練的分類器,研究區分了發生S-S鍵斷裂的吸附與其他類型的吸附。進一步對吸附構型的電子結構分析顯示,負載金屬原子后催化劑與多硫化鋰間的相互作用發生明顯變化,因而使吸附顯著增強,從而減少“穿梭”過程的發生。此外,機器學習訓練出的回歸模型對吸附能也有較好的預測能力,其平均絕對誤差為0.14 eV。基于這一模型,研究預測了上千個吸附構型的吸附能,并利用過電勢的計算給出了相應的火山型曲線。結合可溶性多硫化物的吸附能,研究預測并篩選出數個性能均衡的單原子催化劑。該研究拓寬了單原子催化劑的應用范圍,也為鋰硫電池正極材料的設計提供了新思路。
相關研究成果近日發表在The Journal of Physical Chemistry Letters上,并被選為封面文章。研究工作得到國家自然科學基金、遼寧省自然科學基金材料聯合基金、NSFC-廣東聯合基金(第二期)超級計算科學應用研究專項等的資助。